FDA warns Zoll over MRI ventilator safety lapses
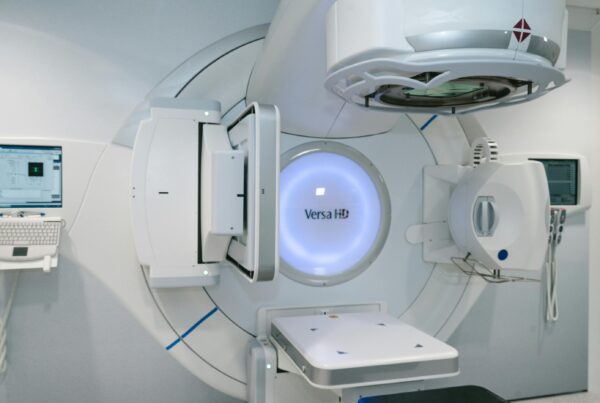
The US Food and Drug Administration has issued a warning letter to Zoll Medical after finding quality and safety lapses tied to its MR conditional ventilator, the device used inside the magnetic resonance imaging suite. Dated April 30, 2026, the letter states that the company failed to properly validate labeling for its 731 ventilator family and was late reporting an incident involving a unit near an MRI scanner.
The inspection behind the action ran from February 27 to April 15, 2025, at Zoll’s plant in Chelmsford, Massachusetts. For any hospital that runs MRI-compatible ventilators in intensive care or imaging departments, the case is a practical reminder that the “MR conditional” label only protects the patient when every condition of use is tested, documented, and followed precisely.
What the FDA found during the inspection
At the heart of the warning is a labeling validation problem. The 731 Series ventilators (AEV, EMV+, and Eagle II models) are indicated for use with both 1.5-tesla and 3-tesla scanners. Yet the FDA determined that MRI compatibility had been tested only on a 3T magnet, with no documented rationale for the absence of 1.5T testing, even though 1.5T is one of the most common field strengths in the global installed base.
The practical fallout shows up in the labeling. The manual told users to place the device behind the 130 Gauss field line for 3T magnets, but it failed to specify how far the ventilator should sit when used with a 1.5T system. Without that limit, staff can position the equipment too close to the magnet, triggering false alarms, malfunction, or unexpected shutdown, precisely when the patient depends on it to breathe.
The agency also flagged a vigilance failure. Zoll learned on June 20, 2024, of a case in which a customer confirmed using the ventilator near a 1.5T system, degrading image quality. The mandatory medical device report (MDR) did not reach the FDA until March 10, 2025, far beyond the 30-calendar-day window required under US law.
Why the Gauss line matters so much
The MRI room is one of the most hazardous environments in the hospital because the main magnetic field is never switched off. It extends outward from the gantry in concentric layers measured in gauss (G): the 5 G line marks the safety boundary for the general public and people with implanted cardiac devices, while higher values such as 130 or 300 G define how close sensitive electronics can operate without interference or ferromagnetic attraction.
Even a device labeled “MR conditional” can contain ferromagnetic parts and is only safe within the specific validated conditions: field strength, spatial gradient, and minimum distance. MRI safety literature reports that repeated exposure above 300 G can degrade ventilator performance. The missing 1.5T limit is therefore not a clerical detail but a gap that can turn a life-support device into a hazard. Readers who follow how the magnetic field changes equipment behavior in the MR suite understand why each tesla counts.
What it means for clinical practice
For the radiographer, the ICU nurse, and the medical physicist on the floor, the lesson is direct: the Zone IV safety protocol must include an explicit check of the Gauss line matching the magnet’s strength before any ventilator approaches the patient. Permanent Gauss-line floor markers and ferromagnetic detectors at the entrance help turn an invisible threat into a visible signal.
The case also underscores the importance of labeling and manual-version control. On June 12, 2024, Zoll had sent customers an Urgent Corrective Action notice asking them to discard old versions of the operator guides. Imaging services should confirm they are using the latest documentation and that staff are trained on the correct distances for both 1.5T and 3T. Such episodes join other recent regulatory alerts, like the FDA recall of Philips fluoroscopy systems, showing that ongoing vigilance is part of the job.
Software, quality system, and next steps
The warning went beyond labeling. The FDA questioned the RescueNet Live software: if it analyzes data from a monitoring device and generates a secondary alarm, rather than merely receiving and displaying alarms, then it qualifies as a medical device and would be adulterated for lacking an approved premarket approval (PMA). The agency also cited failures in design validation procedures, including risk analysis, and in supplier and service-provider controls.
Zoll has a deadline to respond formally with a corrective-action plan. For the sector, the takeaway is that quality systems, vigilance reporting, and MRI safety move together: no label replaces a robust validation process. Professionals working in regulation and training also watch issues such as the push to license imaging technologists, part of the same effort to raise the safety bar in medical imaging. In the coming months, FDA follow-up is expected to confirm the nonconformities are corrected.
Source: AuntMinnie